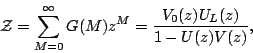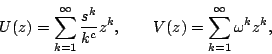 |
Thermal denaturation or melting of double-stranded DNA is the process by which the two strands unbind upon heating. The nature of this transition has been investigated for almost four decades. Experimentally, the transition from bound to unbound is first order.
The early theoretical models [1], which we refer to as Poland-Scheraga (PS) type models, consider the DNA molecule as composed of an alternating sequence of bound and denaturated states (see, e.g., Fig. 1) Typically a bound state is energetically favored over an unbound one, while a denaturated segment (loop) is entropically favored over a bound one. Within the PS type models the segments which compose the chain are assumed to be non-interacting with one another. This assumption considerably simplifies the theoretical treatment and enables one to calculate the resulting free energy. In the past the entropy of the denaturated loops has been evaluated by modelling them either as ideal random walks [2] or as self-avoiding walks [3]. It has been found that within this approach the denaturation transition of DNA is continuous both in two and three dimensions. It becomes first order only above four dimensions.
We have considered analytically the effects of
excluded-volume interaction between the various segments of the
chain [4]. Although we treated this interaction only in an approximate
way, we were able to give some insight into the unbinding mechanism
and on the nature of the transition. Our approach makes use of
recent important results on the entropy of self-avoiding polymer
networks [5,6]. We find that this interaction drives the
transition first order in ![]() , 3 and
, 3 and ![]() dimensions.
dimensions.
The PS model considers two strands, made of monomers, each
representing one persistence length of a single strand (![]() Å). A typical DNA configuration is shown in
Fig. 1. It is made of sequences of bound monomers
separated by denaturated loops. The statistical weight of a bound
sequence of length
Å). A typical DNA configuration is shown in
Fig. 1. It is made of sequences of bound monomers
separated by denaturated loops. The statistical weight of a bound
sequence of length ![]() is
is
![]() , where
, where ![]() is
the temperature. On the other hand the statistical
weight of a denaturated sequence of length
is
the temperature. On the other hand the statistical
weight of a denaturated sequence of length ![]() is given by the
change in entropy due to the added configurations arising from a
loop of length
is given by the
change in entropy due to the added configurations arising from a
loop of length ![]() . For large
. For large ![]() this has the general form
this has the general form ![]() , where
, where ![]() is a non-universal constant and the exponent
is a non-universal constant and the exponent
![]() is determined by the properties of the loop configurations.
For simplicity, we set
is determined by the properties of the loop configurations.
For simplicity, we set ![]() . The model is most easily studied
within the grand canonical ensemble where the total chain length
. The model is most easily studied
within the grand canonical ensemble where the total chain length
![]() is allowed to fluctuate. The grand canonical partition
function,
is allowed to fluctuate. The grand canonical partition
function, ![]() , is given by
, is given by
| (3) |
It is thus clear that the nature of the denaturation transition is
determined by the dependence of ![]() on
on ![]() . The transition takes
place when
. The transition takes
place when ![]() reaches
reaches ![]() . Its
nature is determined by the behaviour of
. Its
nature is determined by the behaviour of ![]() in the vicinity of
in the vicinity of ![]() . This is controlled in turn by the value of the exponent
. This is controlled in turn by the value of the exponent ![]() .
There are three regimes:
.
There are three regimes:
In the above treatment the entropy of a loop of length ![]() was
taken to be of the form
was
taken to be of the form ![]() .
In order to evaluate the exponent
.
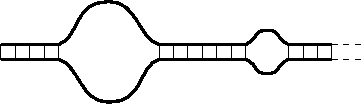
In order to evaluate the exponent ![]() for our configuration, we
use the results obtained by Duplantier et al.[5,6] for
the entropy of general polymer networks. Applying these
results to the relevant topology to
the problem discussed in this work (Fig. 2), we
obtain
the appropriate value of effective exponent
for our configuration, we
use the results obtained by Duplantier et al.[5,6] for
the entropy of general polymer networks. Applying these
results to the relevant topology to
the problem discussed in this work (Fig. 2), we
obtain
the appropriate value of effective exponent ![]() :
:
| (4) |
The exponent ![]() is therefore larger than
is therefore larger than ![]() in both
in both ![]() and
and
![]() , implying
that in these cases the transition from native to denaturated DNA
is first order. This strongly suggests that the transition is also
first-order in
, implying
that in these cases the transition from native to denaturated DNA
is first order. This strongly suggests that the transition is also
first-order in ![]() . This is supported by the Padé and
Padé-Borel approximations in d=3.
. This is supported by the Padé and
Padé-Borel approximations in d=3.
 |